Wang et al: time course of microglia from the rTg4510 mouse model
tau.RmdIn 2018, Wang et al published the transcriptome analysis of mouse microglia, isolated either from the rTg4510 mouse model or wildtype animals. Four age groups of mice (2-, 4-, 6-, and 8-months) were analyzed to capture longitudinal gene expression changes that correspond to varying levels of pathology, from minimal tau accumulation to massive neuronal loss.
The raw RNA-seq data from this study is available from the Short Read Archive, accession SRP172787 Reads were aligned to the mouse reference genome (version GRCm38_p6, Gencode release release_M17) with the STAR aligner (version 2.7.1a) and gene-level abundances were inferred from the BAM files using salmon (version 0.13.1).
A DESeqDataset object with raw counts, sample and gene annotations is available in the tau dataset included in this R package.
data("tau")
dds <- tauDifferential expression analysis with DESeq2
# filtering, just to speed up computations
keep <- rowSums(counts(dds) >= 10) >= min(table(dds$group))
dds <- dds[which(keep & rowData(dds)$gene_type == "protein_coding"), ]
dds <- DESeq(dds)
#> estimating size factors
#> estimating dispersions
#> gene-wise dispersion estimates
#> mean-dispersion relationship
#> final dispersion estimates
#> fitting model and testing
res <- results(dds, contrast = c("group", "rTg4510_2", "WT_2"))
res <- res[order(res$pvalue), ]
top_hits <- head(row.names(res))
data.frame(
symbol = rowData(dds)[top_hits, c("symbol")],
res[top_hits, ]
)
#> symbol baseMean log2FoldChange lfcSE stat
#> ENSMUSG00000029304 Spp1 2616.790 2.359567 0.1723853 13.687748
#> ENSMUSG00000004552 Ctse 4242.904 1.549891 0.1490390 10.399229
#> ENSMUSG00000016494 Cd34 5061.259 1.137964 0.1160378 9.806840
#> ENSMUSG00000015396 Cd83 17178.022 1.797699 0.1855271 9.689683
#> ENSMUSG00000033350 Chst2 1900.333 1.546347 0.1605566 9.631164
#> ENSMUSG00000068129 Cst7 1027.432 1.769383 0.1860918 9.508118
#> pvalue padj
#> ENSMUSG00000029304 1.201765e-42 1.481056e-38
#> ENSMUSG00000004552 2.499468e-25 1.540172e-21
#> ENSMUSG00000016494 1.052112e-22 4.322078e-19
#> ENSMUSG00000015396 3.335609e-22 1.027701e-18
#> ENSMUSG00000033350 5.905648e-22 1.455624e-18
#> ENSMUSG00000068129 1.941432e-21 3.987701e-18
DESeq2::plotMA(res)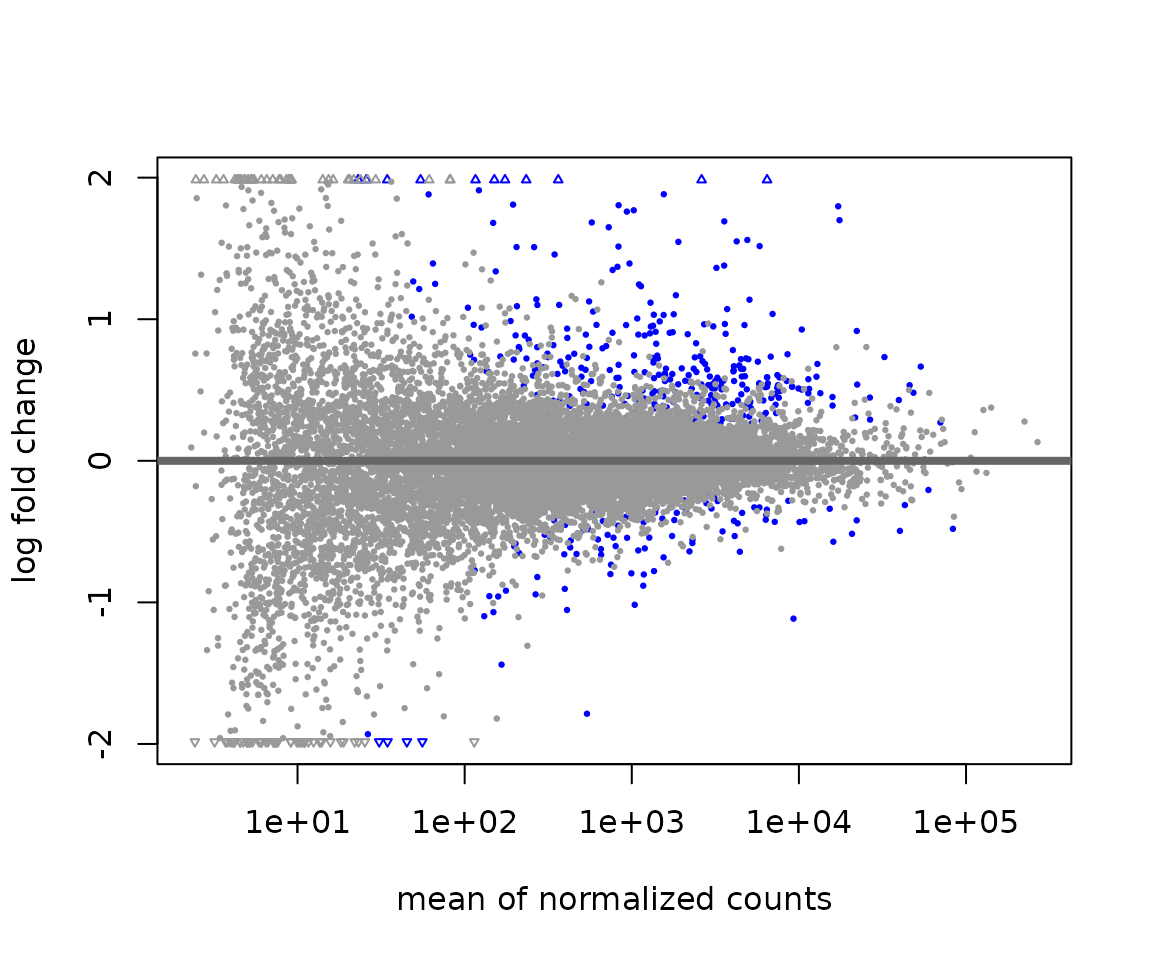
plotCounts(dds, gene = top_hits[1], intgroup="group",
main = rowData(dds)[top_hits[1], "symbol"])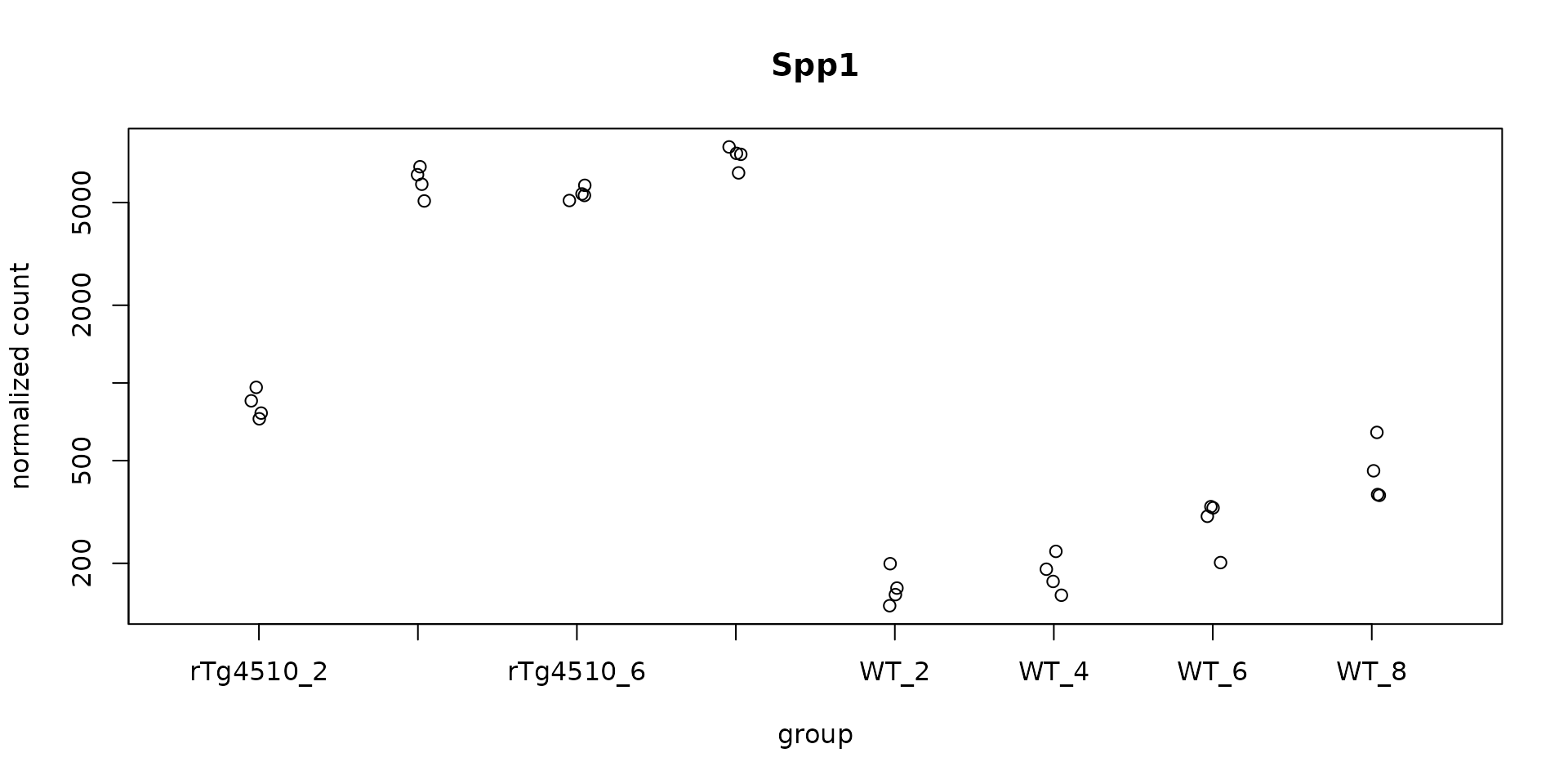
Limma / Voom analysis
Alternatively, we can perform the same analysis using the limma/voom framework, specifically the voomLmFit() function from the edgeR Bioconductor package.
design <- model.matrix(~ 0 + group, data = colData(dds))
colnames(design) <- sub("group", "", colnames(design))
keep <- filterByExpr(dds, design = design)
fit <- voomLmFit(
counts = dds[keep & rowData(dds)$gene_type == "protein_coding", ],
design = design,
plot = TRUE,
sample.weights = TRUE)
#> First sample weights (min/max) 0.06777076/1.83011791
#> Final sample weights (min/max) 0.06486057/1.85889345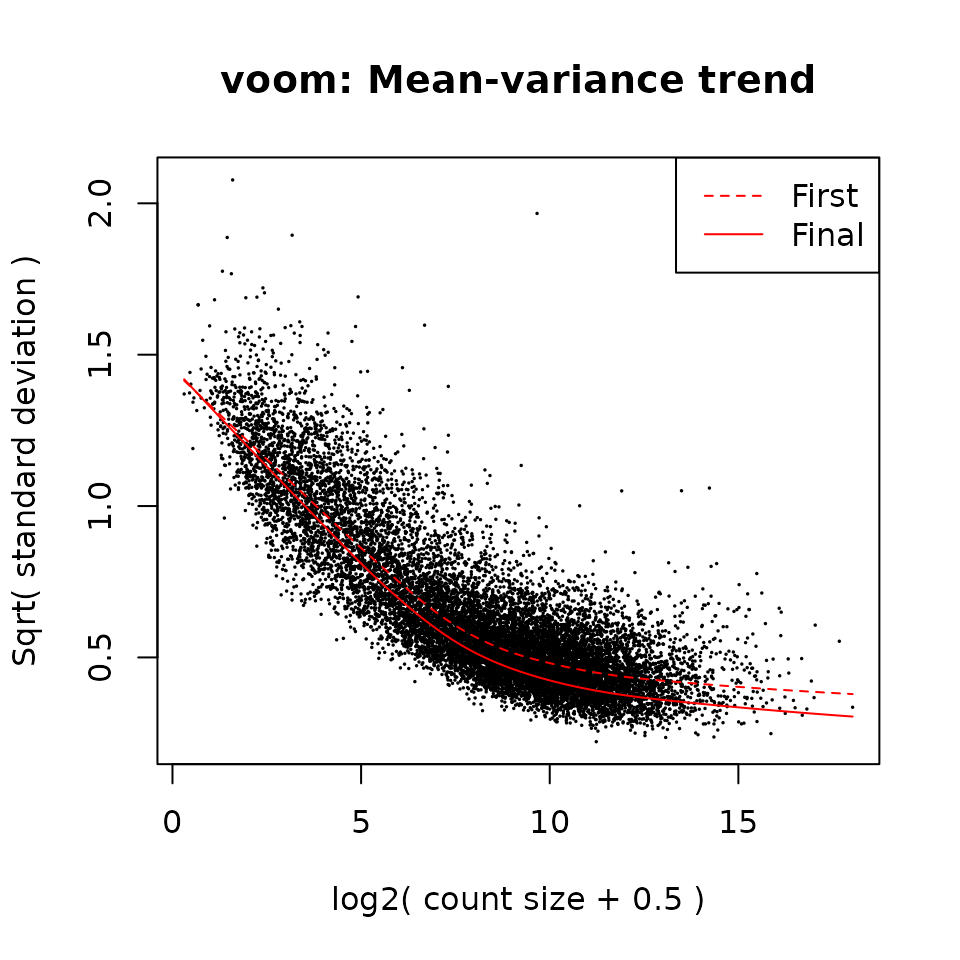
contrasts = makeContrasts(
two_months = "rTg4510_2 - WT_2",
four_months = "rTg4510_4 - WT_4",
six_months = "rTg4510_6 - WT_6",
eight_months = "rTg4510_8 - WT_8",
levels = design
)
fit2 <- contrasts.fit(fit, contrasts = contrasts)
fit2 <- eBayes(fit2)
topTable(fit2, coef = "two_months",
p.value = 0.05)[, c("symbol", "logFC", "P.Value", "adj.P.Val")]
#> symbol logFC P.Value adj.P.Val
#> ENSMUSG00000016494 Cd34 1.0841155 4.008578e-15 2.845289e-11
#> ENSMUSG00000029304 Spp1 2.3735606 3.105941e-15 2.845289e-11
#> ENSMUSG00000015396 Cd83 1.8632546 4.146542e-14 1.962144e-10
#> ENSMUSG00000015568 Lpl 1.5345569 5.161900e-13 1.591677e-09
#> ENSMUSG00000004552 Ctse 1.5125064 5.606074e-13 1.591677e-09
#> ENSMUSG00000031210 Gpr165 -1.1588124 1.174731e-12 2.779414e-09
#> ENSMUSG00000033350 Chst2 1.5757559 1.517534e-12 3.077559e-09
#> ENSMUSG00000018927 Ccl6 0.8710038 4.700783e-12 8.341539e-09
#> ENSMUSG00000032076 Cadm1 0.6636275 6.182402e-12 8.698938e-09
#> ENSMUSG00000014599 Csf1 2.0160775 6.321838e-12 8.698938e-09
sessionInfo()
#> R version 4.2.1 (2022-06-23)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 20.04.5 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
#> LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices datasets utils methods
#> [8] base
#>
#> other attached packages:
#> [1] rnaseqExamples_0.0.0.9000 edgeR_3.38.4
#> [3] limma_3.52.4 DESeq2_1.36.0
#> [5] SummarizedExperiment_1.26.1 Biobase_2.56.0
#> [7] MatrixGenerics_1.8.1 matrixStats_0.62.0
#> [9] GenomicRanges_1.48.0 GenomeInfoDb_1.32.4
#> [11] IRanges_2.30.1 S4Vectors_0.34.0
#> [13] BiocGenerics_0.42.0
#>
#> loaded via a namespace (and not attached):
#> [1] httr_1.4.4 sass_0.4.2 bit64_4.0.5
#> [4] jsonlite_1.8.2 splines_4.2.1 bslib_0.4.0
#> [7] highr_0.9 blob_1.2.3 renv_0.16.0
#> [10] GenomeInfoDbData_1.2.8 yaml_2.3.6 pillar_1.8.1
#> [13] RSQLite_2.2.18 lattice_0.20-45 glue_1.6.2
#> [16] digest_0.6.30 RColorBrewer_1.1-3 XVector_0.36.0
#> [19] colorspace_2.0-3 htmltools_0.5.3 Matrix_1.4-1
#> [22] pkgconfig_2.0.3 XML_3.99-0.11 genefilter_1.78.0
#> [25] zlibbioc_1.42.0 purrr_0.3.5 xtable_1.8-4
#> [28] scales_1.2.1 BiocParallel_1.30.4 tibble_3.1.8
#> [31] annotate_1.74.0 KEGGREST_1.36.3 ggplot2_3.3.6
#> [34] cachem_1.0.6 cli_3.4.1 survival_3.3-1
#> [37] magrittr_2.0.3 crayon_1.5.2 memoise_2.0.1
#> [40] evaluate_0.17 fansi_1.0.3 fs_1.5.2
#> [43] textshaping_0.3.6 tools_4.2.1 lifecycle_1.0.3
#> [46] stringr_1.4.1 locfit_1.5-9.6 munsell_0.5.0
#> [49] DelayedArray_0.22.0 AnnotationDbi_1.58.0 Biostrings_2.64.1
#> [52] compiler_4.2.1 pkgdown_2.0.6 jquerylib_0.1.4
#> [55] systemfonts_1.0.4 rlang_1.0.6 grid_4.2.1
#> [58] RCurl_1.98-1.9 bitops_1.0-7 rmarkdown_2.17
#> [61] gtable_0.3.1 codetools_0.2-18 DBI_1.1.3
#> [64] R6_2.5.1 knitr_1.40 utf8_1.2.2
#> [67] fastmap_1.1.0 bit_4.0.4 rprojroot_2.0.3
#> [70] ragg_1.2.3 desc_1.4.2 stringi_1.7.8
#> [73] parallel_4.2.1 Rcpp_1.0.9 vctrs_0.4.2
#> [76] geneplotter_1.74.0 png_0.1-7 xfun_0.34