Naguib et al: Global effects of SUPT4H1 RNAi on gene expression.
rnai.RmdThe SUPT4H1 protein is a highly conserved transcription elongation factor that makes up part of the RNA polymerase II complex. To investigate the role of SUPT4H1 in transcriptional regulation, Naguib et al depleted SUPT4H1 transcripts by RNA interference in HEK293 cells.
Because loss of SUPT4H1 leads to a global decrease in RNA polymerase II dependent transcription, the authors included spike-ins to allow for normalization independent of total gene counts.
The processed RNA-seq data from this study is available from the NCBI GEO (accession GSE116267) Reads were aligned to the human reference genome (version GRCh38_p10, including sequences of 92 ERCC spike-in transcripts) with the STAR aligner (version 2.5.3a) and gene-level abundances were inferred using salmon (version version 0.9.1).
A DESeqDataset object with raw counts, sample and gene annotations from one of the exoeriment published by Naguib et al, RNAi in HEK293 cells, is available in the `rnai`` dataset included in this R package.
data("rnai")
dds <- rnaiDifferential expression analysis with DESeq2
# filtering, just to speed up computations
keep <- rowSums(counts(dds) >= 10) >= min(table(dds$group))
dds <- dds[which(
(keep & rowData(dds)$gene_type == "protein_coding") |
(rowData(dds)$spikein)), ]
dds <- estimateSizeFactors(dds, controlGenes = which(rowData(dds)$spikein))
dds <- DESeq(dds)
#> using pre-existing size factors
#> estimating dispersions
#> gene-wise dispersion estimates
#> mean-dispersion relationship
#> final dispersion estimates
#> fitting model and testing
res <- results(dds,
contrast = c("group", "RNAi", "Ctrl"),
alpha = 0.1) # only used to optimize IHW
table(res$padj < 0.01 & abs(res$log2FoldChange) > 0.5)
#>
#> FALSE TRUE
#> 1918 11545As reported in the original manuscript, the vast majority of genes shows decreased transcription following SUPT4H1 RNAi treatment.
hist(res$log2FoldChange, breaks = 100, xlim = c(-2, 2),
xlab = "RNAi vs Ctrl (log2FC)", main = "RNAi vs Ctrl")
abline(v = 0, lwd = 2, col = "red")
title(sub = "DESeq2")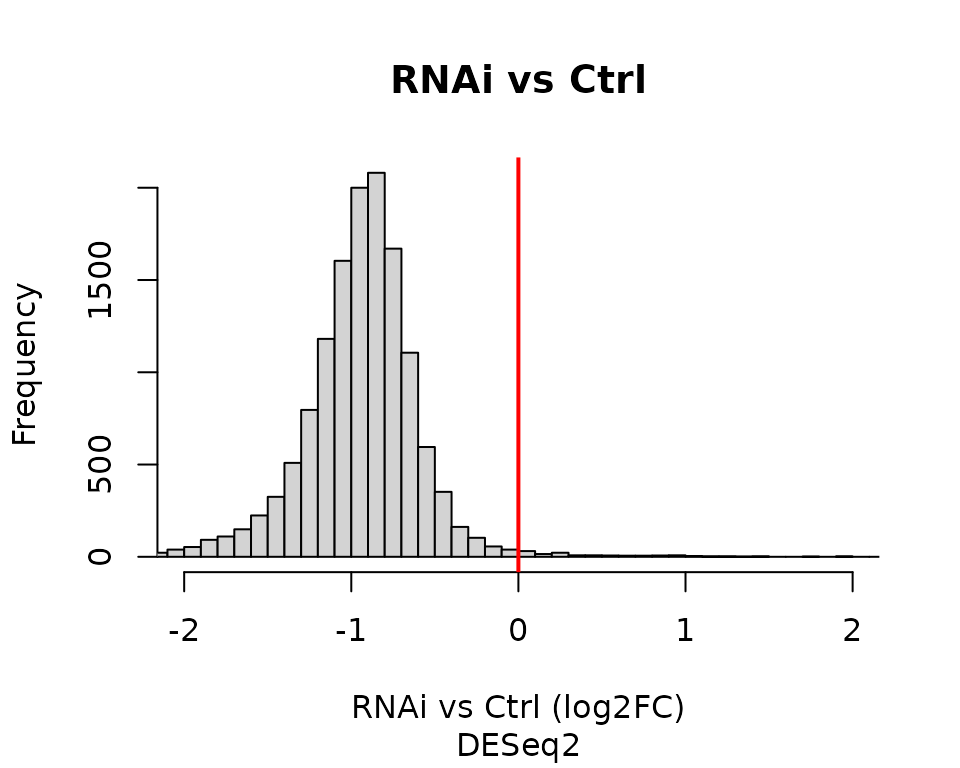
Limma / Voom analysis
Alternatively, we can perform the same analysis using the limma/voom framework, specifically the voomLmFit() function from the edgeR Bioconductor package.
design <- model.matrix(~ 0 + group, data = colData(dds))
colnames(design) <- sub("group", "", colnames(design))
keep <- filterByExpr(dds, design = design)
fit <- voomLmFit(
counts = dds,
design = design,
plot = FALSE,
# use only the spike-ins to calculate library size
lib.size = colSums(counts(dds)[rowData(dds)$spikein, ]),
sample.weights = TRUE)
#> First sample weights (min/max) 0.206246/4.865506
#> Final sample weights (min/max) 0.2023536/4.7824662
contrasts = makeContrasts(
"RNAi - Ctrl",
levels = design
)
fit2 <- contrasts.fit(fit, contrasts = contrasts)
fit2 <- eBayes(fit2)
tt <- topTable(fit2, n = Inf)[, c("symbol", "logFC", "P.Value", "adj.P.Val")]
hist(tt$logFC, breaks = 100, xlim = c(-2, 2), xlab = "RNAi vs Ctrl (log2FC)",
main = "SUPT4H1 RNAi vs Ctrl")
abline(v = 0, lwd = 2, col = "red")
title(sub = "limma/voom")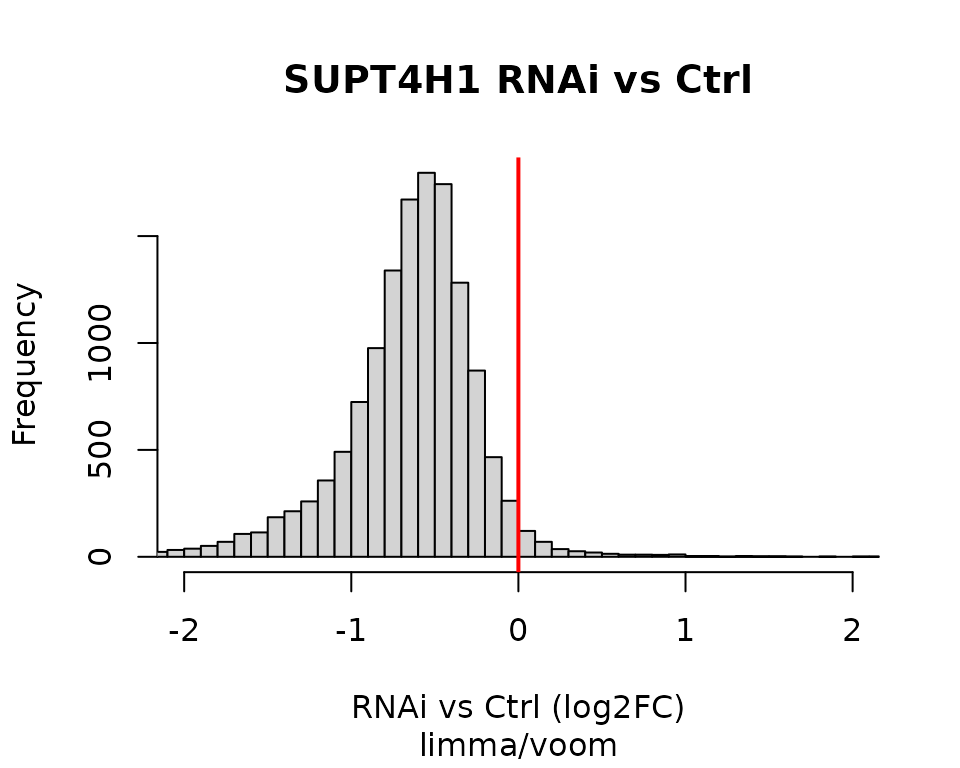
sessionInfo()
#> R version 4.2.1 (2022-06-23)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 20.04.5 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
#> LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices datasets utils methods
#> [8] base
#>
#> other attached packages:
#> [1] DESeq2_1.36.0 SummarizedExperiment_1.26.1
#> [3] Biobase_2.56.0 MatrixGenerics_1.8.1
#> [5] matrixStats_0.62.0 GenomicRanges_1.48.0
#> [7] GenomeInfoDb_1.32.4 IRanges_2.30.1
#> [9] S4Vectors_0.34.0 BiocGenerics_0.42.0
#> [11] rnaseqExamples_0.0.0.9000 edgeR_3.38.4
#> [13] limma_3.52.4
#>
#> loaded via a namespace (and not attached):
#> [1] httr_1.4.4 sass_0.4.2 bit64_4.0.5
#> [4] jsonlite_1.8.2 splines_4.2.1 bslib_0.4.0
#> [7] highr_0.9 blob_1.2.3 renv_0.16.0
#> [10] GenomeInfoDbData_1.2.8 yaml_2.3.6 pillar_1.8.1
#> [13] RSQLite_2.2.18 lattice_0.20-45 glue_1.6.2
#> [16] digest_0.6.30 RColorBrewer_1.1-3 XVector_0.36.0
#> [19] colorspace_2.0-3 htmltools_0.5.3 Matrix_1.4-1
#> [22] pkgconfig_2.0.3 XML_3.99-0.11 genefilter_1.78.0
#> [25] zlibbioc_1.42.0 purrr_0.3.5 xtable_1.8-4
#> [28] scales_1.2.1 BiocParallel_1.30.4 tibble_3.1.8
#> [31] annotate_1.74.0 KEGGREST_1.36.3 ggplot2_3.3.6
#> [34] cachem_1.0.6 cli_3.4.1 survival_3.3-1
#> [37] magrittr_2.0.3 crayon_1.5.2 memoise_2.0.1
#> [40] evaluate_0.17 fansi_1.0.3 fs_1.5.2
#> [43] textshaping_0.3.6 tools_4.2.1 lifecycle_1.0.3
#> [46] stringr_1.4.1 munsell_0.5.0 locfit_1.5-9.6
#> [49] DelayedArray_0.22.0 AnnotationDbi_1.58.0 Biostrings_2.64.1
#> [52] compiler_4.2.1 pkgdown_2.0.6 jquerylib_0.1.4
#> [55] systemfonts_1.0.4 rlang_1.0.6 grid_4.2.1
#> [58] RCurl_1.98-1.9 bitops_1.0-7 rmarkdown_2.17
#> [61] gtable_0.3.1 codetools_0.2-18 DBI_1.1.3
#> [64] R6_2.5.1 knitr_1.40 utf8_1.2.2
#> [67] fastmap_1.1.0 bit_4.0.4 rprojroot_2.0.3
#> [70] ragg_1.2.3 desc_1.4.2 stringi_1.7.8
#> [73] parallel_4.2.1 Rcpp_1.0.9 vctrs_0.4.2
#> [76] geneplotter_1.74.0 png_0.1-7 xfun_0.34